Images
QuPath is software for image analysis. This section gives a brief overview of digital images, and the techniques and concepts needed to analyze them using QuPath.
Tip
For a more extensive introduction to images and bioimage analysis concepts, see [Introduction to Bioimage Analysis].
Introducing pixels
Visualizing pixels
Digital images are composed of pixels, which is a term derived from picture element.
When zooming in a lot, each pixel is typically visualized as a small square of a certain color.
However, this is just a visualization. As far as the computer is concerned, each pixel is really just a number and the full image is a 2D matrix of these numbers: the pixel values. The values just happen to be displayed using colors for our benefit to aid interpretation.
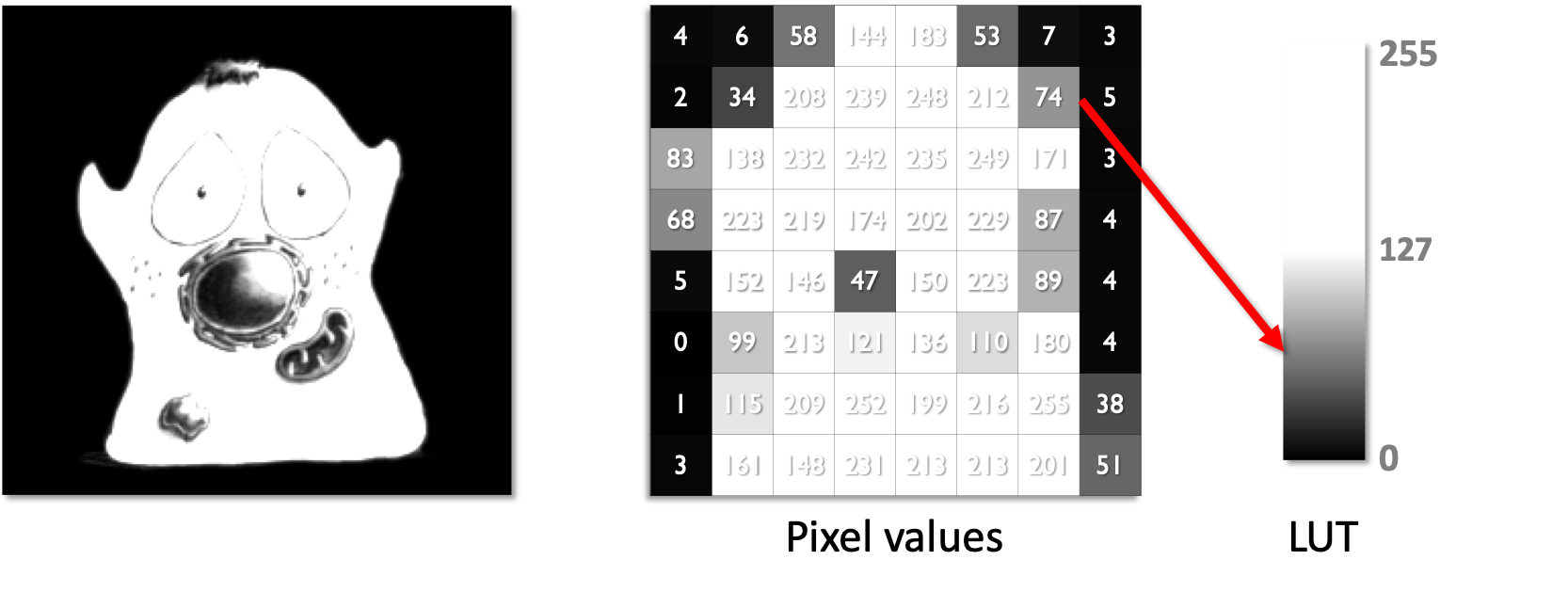
Illustration an image as it is normally visualized (left) and how the structures apparent within the image are reflected in the underlying pixel values that make up the image (right). In science, the pixel values are crucial - how they are displayed is not.
Choosing colors
The color used to display each pixel is defined by a lookup table (LUT).
The figure below shows an original image (left) alongside a lower-resolution version so that individual pixel values can be easily seen (middle). Beside these, a LUT is shown (right) containing 256 shades of gray, corresponding to the values 0-255 - which also represents the range of pixel values within the image. For each pixel in both images, the pixel value is used to index the LUT and find out which color to display for that pixel.

By separating the display color from the pixel value using LUTs, it becomes possible to change the brightness/contrast of an image without changing the underlying pixel values - simply by changing the LUT.
This is crucial, because the pixel values are the raw data in scientific imaging. These need to be preserved unchanged even if we visualize them in different ways.
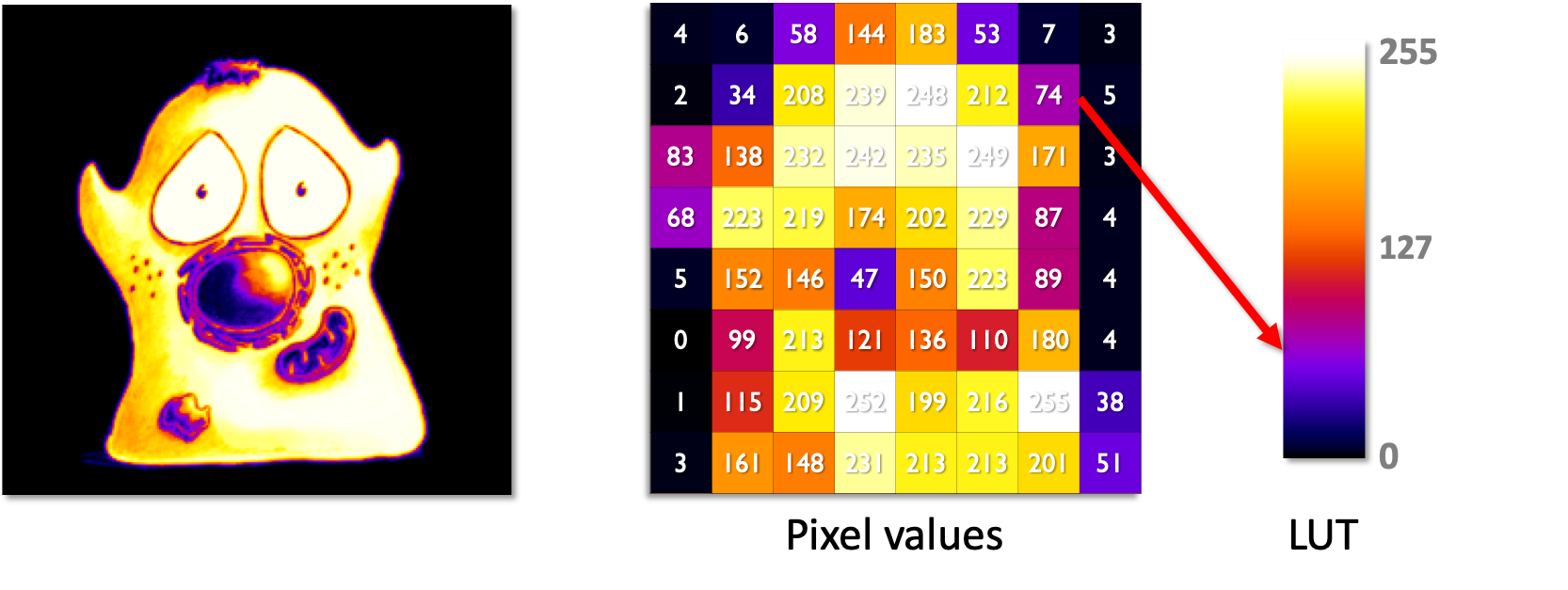
Of course, having separated pixel values from the colors used to display them, there is no reason to restrict ourselves to only shades of gray. Other LUTs can be used to add other colors.
Warning
Scientific image analysis is not photo editing!
The separation between the values and how they are displayed is so important because scientific measurements must generally be made based on the raw data - and any deviations from that must be tractable and justifiable. If a simple step like adjusting the brightness would modify the pixel values this could compromise all later measurements.
It is important to be aware that most common image/photo editing software applications don’t care about preserving pixel values. Only the appearance really matters. Therefore if you adjust the brightness/contrast of an image in such software you can easily make the image unsuitable for later quantitative analysis by compromising the data.
Interpolation issues
Pixels within images do not have to be visualized as little discrete squares. In fact, a famous paper A Pixel is Not A Little Square argues against relying exclusively on this model.
An alternative visualization might use a smoother interpolation between pixel values when zoomed in. One example is bilinear interpolation, available in QuPath under .

Image viewed in QuPath using the default (nearest-neighbor, ‘square’) interpolation (left) and also bilinear interpolation (right).
However, it is generally advisable to turn bilinear interpolation off, since the default ‘square’ interpolation gives a more accurate depiction of the level of detail present in the image. The blocky appearance serves as a useful warning that our images do not contain perfect detail.
From pixels to microns
How big is a pixel?
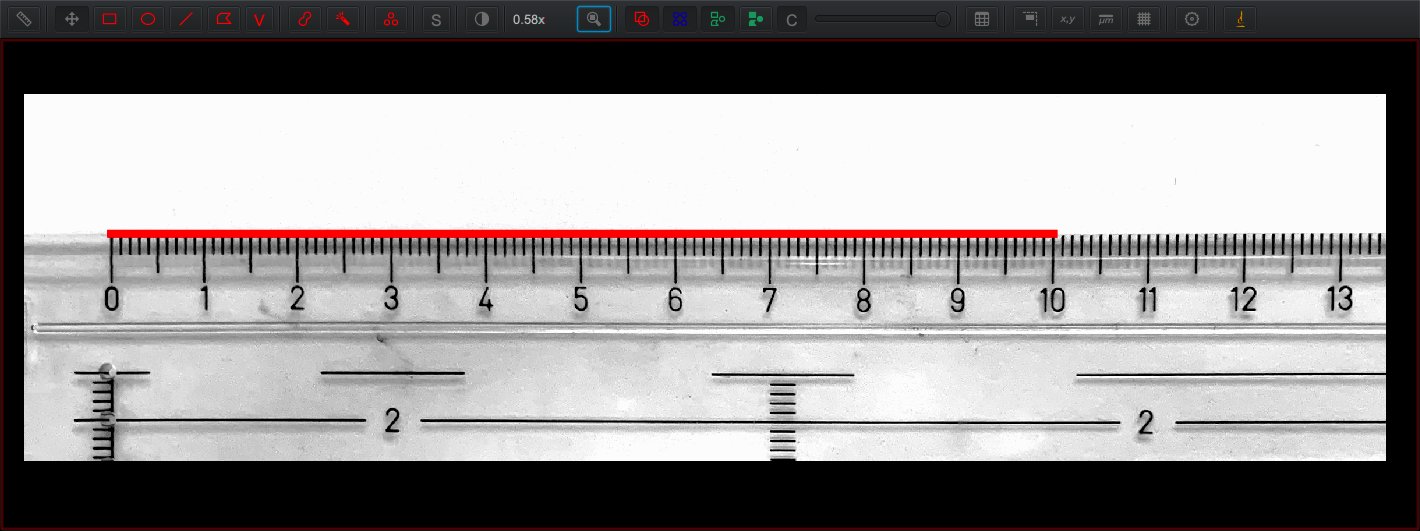
Although pixels don’t really have a ‘size’ (just a numeric value), they can compose images of things that exist in reality and that do have a size. It can therefore be helpful to think (informally) about ‘pixel size’ when considering how to convert measurements made within an image to physical units.
For example, consider the photograph below showing a ruler. The red horizontal line spans 1618 pixels in length (which can be determined just by counting them) and corresponds to 10 cm in the image. We can then infer that the ‘pixel width’ is approximately 10/1618 = 0.0061 cm, or 61 µm.
Tip
Usually the pixel width and height are identical. However for some images this is not the case; depending upon the detector properties, there can be differences.
Where do pixel sizes come from?
Pixel sizes are often written into the files saved during image acquisition - at least if the default file format is selected. If QuPath (or rather one of its designated image reading libraries) is able to parse this information it will be shown under the Image tab and used within the software for measurements.
Warning
Pixel size information is not guaranteed to be a) present, or b) correct within a file! This can cause problems and even wrong results.
For example, most generic image formats (e.g. jpeg, png) will typically not preserve the required pixel size. Others (e.g. tiff) might preserve the pixel size correctly, or it might instead use a ‘dots per inch’ value that really relates to how the image should be printed - and not the sizes of structures within it.
Therefore one should always sanity-check pixel sizes when these are used for analysis.
Pixel size, resolution & magnification
The pixel size relates to the resolution within the detail: informally, the amount of detail that can be seen. As a rule of thumb, a smaller pixel size means more detail is available.
Another way this is often expressed is using the magnification of the objective lens used during image acquisition. Therefore, for example, an image scanned at x40 magnification may have a pixel size of 0.25 µm, while an image from the same scanner acquired at x20 magnification could have a pixel size of 0.5 µm - indicating that higher magnfication provides smaller pixels and more detail.
However, it is important to recognize that the exact relationship between magnification and pixel size is highly scanner/microscope-dependent. In other words, a x20 image from one scanner might have a quite different pixel size compared to a x20 image from another scanner.
Examples of the same slide scanned with an Aperio (left) and a Trestle (right) scanner.
Whole slide images from the OpenSlide freely distributable test data.
Tip
In practice, magnification is then not terribly meaningful: the pixel size is the crucial value needed to make measurements in physical units, and to compare the resolution across images.
Limits of light
One might assume that it is possible to get more and more detail in an image by decreasing the pixel size, and thereby increasing the resolution.
In practice, for light microscopy one quickly runs up against the diffraction limit, which has the practical impact of making small things look blurry.
The details are not essential right now (for a more thorough introduction see Blur and the PSF) but, very crudely, this means that anything smaller than a few hundred nm will appear at least to be at least this large when imaged with a (conventional) light microscope, and any structures closer than this limit can merge and appear as one.
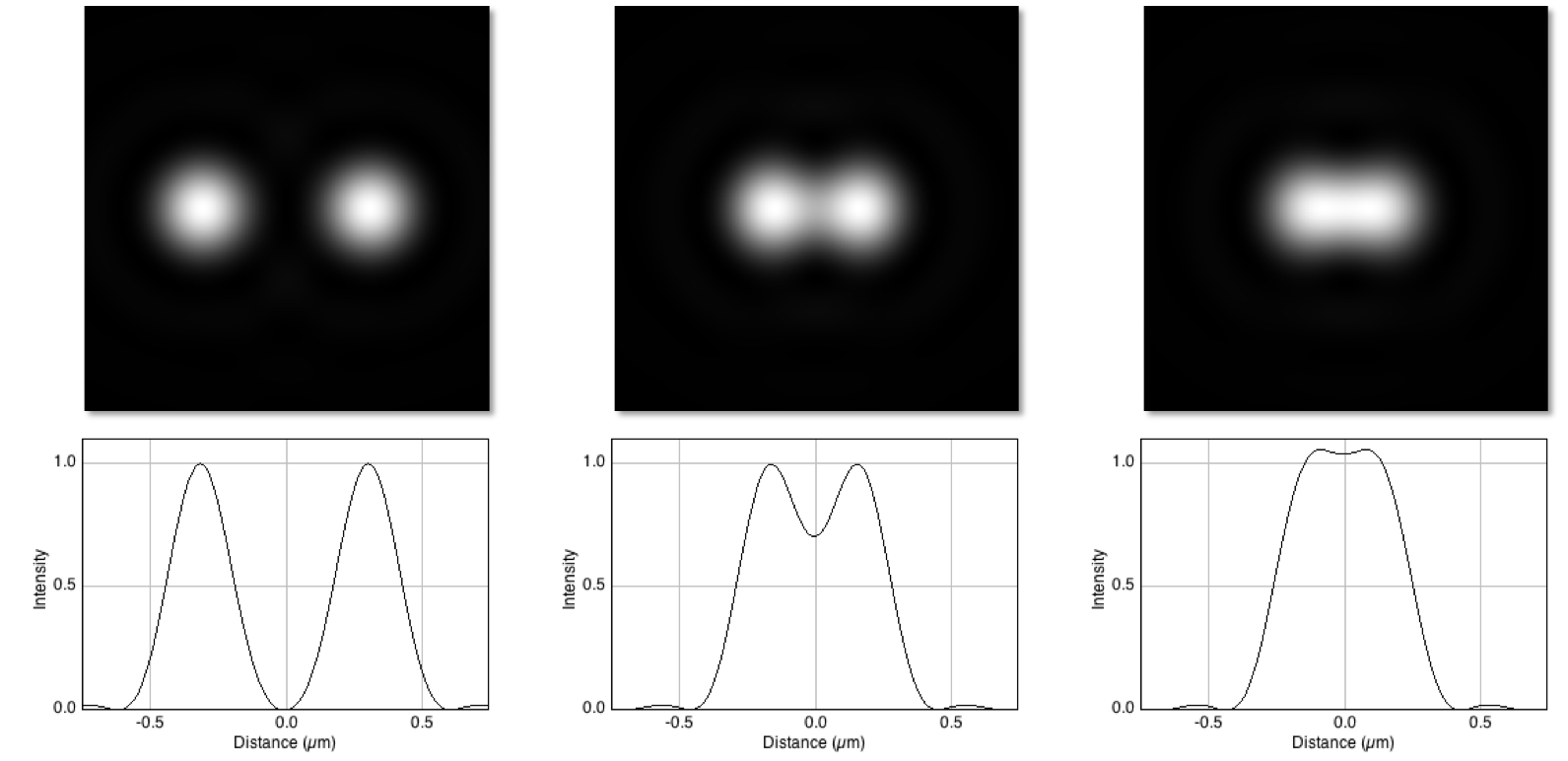
Illustration of how the diffraction limit can make small nearby structures appear to merge.
(Top) Diffraction-limited (i.e. blurry) images of two identical point-like structures, separated by different distances. (Bottom) Plots of the pixel values across the centre of each corresponding image.
Channels & colors
RGB images
Many images - including most brightfield images in QuPath - have three color channels: red, green and blue.
One way to think of such RGB images is that each pixel effectively has three values rather than one, and these represent the amounts of red, green and blue that should generally be used to display the pixel.

Red, green and blue channels combining to create an RGB image (here, a brightfield H&E scan).
These red, green and blue values are depicted in the bottom right corner when moving the cursor over an image in QuPath.
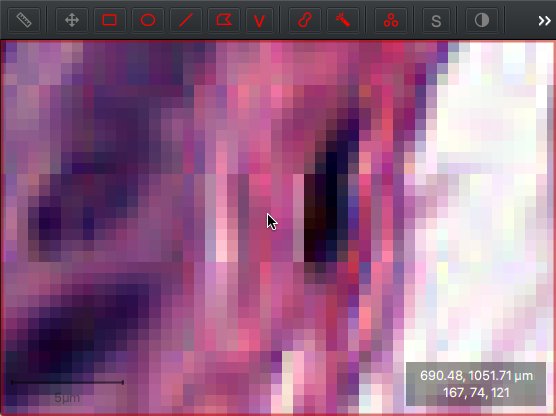
Red, green and blue values shown below the cursor location.
Typically, a pixel with RGB values 0,0,0 would be shown as black and 255,255,255 would be white. The 167,74,121 shown in the screenshot indicates the pink pixel under the cursor is ‘mostly red, with quite a bit of blue and not a lot of green’.
However, the lookup table concept remains: we effectively have three channels that can be treated as separate images, each with its own LUT to map pixel values to colors. We can adjust the brightness/contrast by changing the LUTs independently or all together. The final colors we see are a mixture of the the red, green and blue components calculated for each pixel.

RGB image as above, with brightness/contrast adjustments applied to each channel.
Multichannel images
Not every color image is an RGB image, and not every image has either one or three channels. Multiplexed images may have up to 40 channels or more. QuPath supports these images too.
enables you to view the names of each channel, toggle channels on or off (for display), double-click on a channel to change the color used to visualize it, and adjust brightness/contrast for each channel independently.
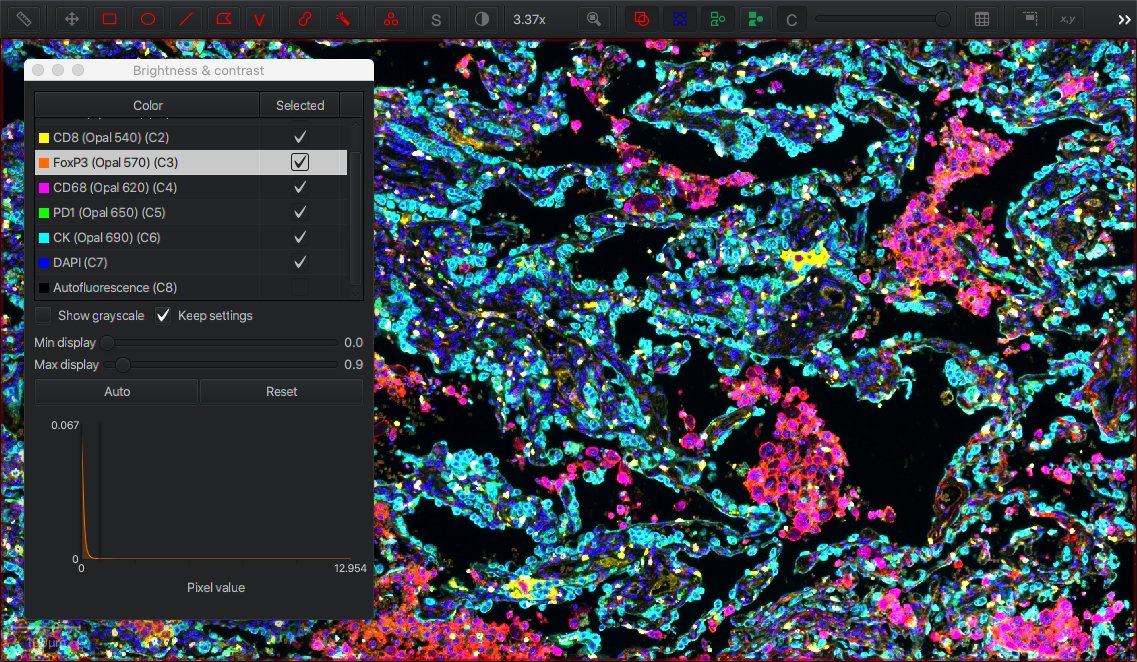
Furthermore, makes it possible to see multiple channels simultaneously, side-by-side.
Tip
Press and hold Shift to pause the viewer, or right click on the channel viewer to customize its display.
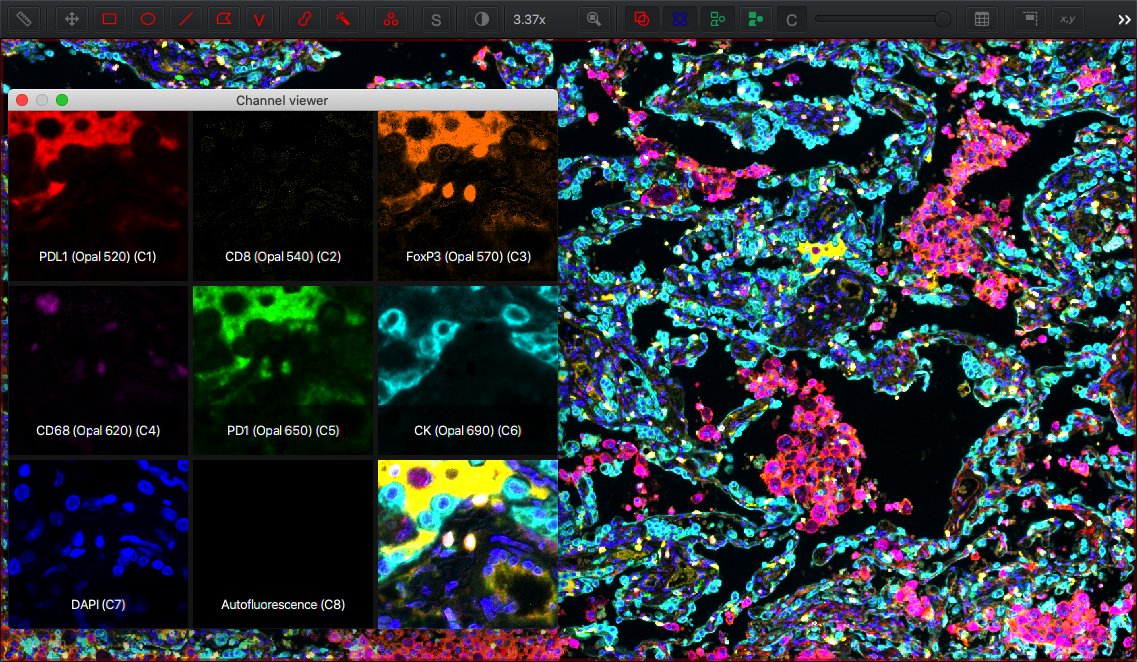
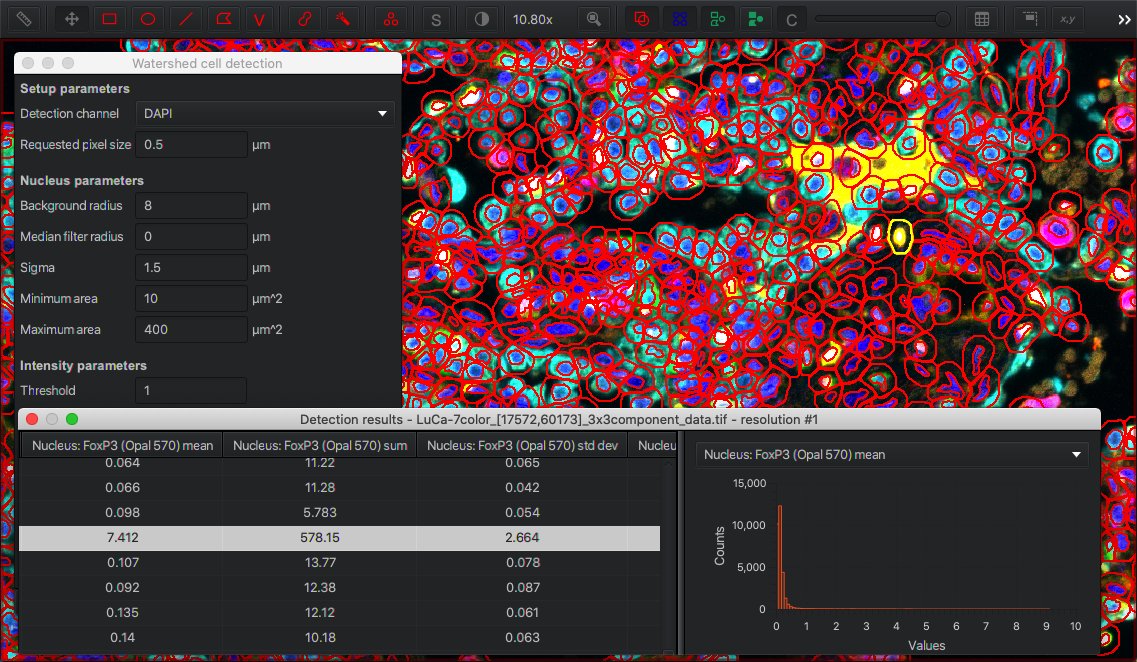
Cell detection in QuPath enables you to select which channel contains nuclear staining (for detection), and will then subsequently measure intensity values within different cell compartments for all channels.
Color deconvolution
Often, multichannel images are much more suitable for quantitative analysis than the brightfield images commonly encountered in pathology. It is certainly helpful that each channel of a fluorescence image (usually) corresponds to something that should be independently measured.
Nevertheless, brightfield images often do need to be analyzed and this requires additional effort (and caveats).
Recall that brightfield images are typically RGB, so each channel corresponds to an amount of red, green or blue light - but not the actual stain used (often hematoxylin, eosin or DAB). In such cases, one may try to computationally recover some estimate of staining intensity from the RGB values.
Probably the most common technique for doing so is color deconvolution, as described by Ruifrok and Johnston. In its original form, color deconvolution can separate up to three stains from an RGB image. Achieving this requires three pieces of information:
The RGB values in the background (which would be
255,255,255if ‘perfectly white’).Three stain vectors characterizing the colors for each stain.
Tip
If only two stains should be separated, a third vector can automatically be generated as orthogonal to the first two vectors. QuPath will do this automatically if needed.
Tip
For brightfield RGB images with two stains only, the command gives a computer-assisted way to determine the stain vectors.
Caution
Color deconvolution & quantification
The quantitative nature of the recovered values is dubious, especially with DAB. However, with cautious interpretation, color deconvolution is a very useful, pragmatic technique to convert RGB values into something a bit closer to what one wishes to assess. It is especially useful when combined with thresholding to distinguish broad categories (e.g. stained vs. unstained).
Tip
Gabriel Landini has written a popular color deconvolution plugin for ImageJ, and describes some important considerations and caveats to keep in mind when using the technique on his website.
Multidimensional images
z-stacks & time series
QuPath has some limited support for z-stacks (images acquired by sampling planes at different depths of focus) and time series (images acquired by sampling at different time points).
Essentially, sliders appear when such a multidimensional image is opened to make it possible to navigate between separate image planes. Objects (e.g. annotations, cells) also should remember which plane they belong to.
For more sophisticated multidimensional image analysis you might want to turn to other software, such as Fiji.